
Conceptualizing a Genomics Software Institute (GSI).
Microbial ecology has been enhanced tremendously by the continued ‘omics revolution, bringing half the world’s biomass and most of its biodiversity into analytical view for the primary time; certainly, it feels virtually just like the invention of the microscope and the invention of the brand new world on the similar time.
With main microbial ecology analysis efforts accumulating prodigious portions of sequence, protein, and metabolite information, we are actually poised to deal with environmental microbial analysis at macro scales, and to start to characterize and perceive the scale of microbial biodiversity on the planet.
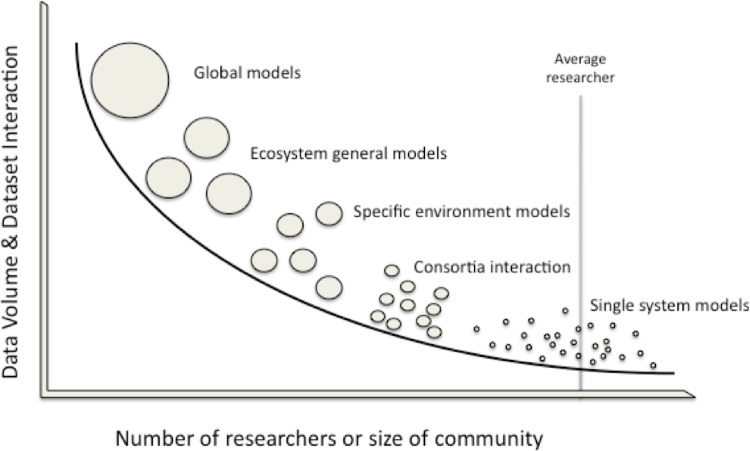
What is at the moment impeding progress is the necessity for a framework inside which the analysis neighborhood can develop, alternate and focus on predictive ecosystem fashions that describe the biodiversity and useful interactions.
Such a framework should embody information and metadata transparency and interoperation; information and outcomes validation, curation, and search; software programming interfaces for modeling and evaluation instruments; and human and technical processes and providers crucial to make sure broad adoption.
Here we focus on the necessity for centered neighborhood interplay to enhance and deepen established neighborhood efforts, starting with the Genomic Standards Consortium (GSC), to create a science-driven strategic plan for a Genomic Software Institute (GSI).
CHEK2 genomic and proteomic analyses reveal genetic inactivation or endogenous activation throughout the 60 cell traces of the US National Cancer Institute.
CHEK2 encodes a serine/threonine kinase (Chk2) activated by ATM in response to DNA double-strand breaks. On the one hand, CHEK2 has been described as a tumor suppressor with proapoptotic, cell-cycle checkpoint and mitotic features.
On the opposite hand, Chk2 can also be generally activated (phosphorylated at T68) in cancers and precancerous lesions. Here, we report an intensive characterization of CHEK2 throughout the panel of 60 established most cancers cell traces from the NCI Anticancer Screen (the NCI-60) utilizing genomic and proteomic analyses, together with exon-specific mRNA expression, DNA copy-number variation (CNV) by aCGH, exome sequencing, in addition to western blot analyses for whole and activated (pT68-Chk2) Chk2.
We present that the excessive heterogeneity of Chk2 ranges in most cancers cells is primarily as a consequence of its inactivation (owing to low gene expression, various splicing, level mutations, copy-number alterations and untimely truncation) or discount of protein ranges.
Moreover, we observe that a vital share of most cancers cells (12% of the NCI-60 and HeLa cells) present excessive endogenous Chk2 activation, which is all the time related to p53 inactivation, and which is accompanied by downregulation of the Fanconi anemia and homologous recombination pathways. We additionally report the presence of activated Chk2 (pT68-Chk2) together with histone γ-H2AX in centrosomes.